Preprocessing
Data analysis - EDA
Dataset preprocessing examples
from mlearner.preprocessing import DataAnalyst
import numpy as np
import pandas as pd
from mlearner.preprocessing import DataAnalyst
%matplotlib inline
Load data in base_preprocess class
filename = "mlearner/data/data/breast-cancer-wisconsin-data.txt"
dataset = DataAnalyst.load_data(filename, sep=",")
dataset.data.head(5)
| Sample code number | Clump Thickness | Uniformity of Cell Size | Uniformity of Cell Shape | Marginal Adhesion | Single Epithelial Cell Size | Bare Nuclei | Bland Chromatin | Normal Nucleoli | Mitoses | Class | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1000025 | 5 | 1 | 1 | 1 | 2 | 1 | 3 | 1 | 1 | 2 |
| 1 | 1002945 | 5 | 4 | 4 | 5 | 7 | 10 | 3 | 2 | 1 | 2 |
| 2 | 1015425 | 3 | 1 | 1 | 1 | 2 | 2 | 3 | 1 | 1 | 2 |
| 3 | 1016277 | 6 | 8 | 8 | 1 | 3 | 4 | 3 | 7 | 1 | 2 |
| 4 | 1017023 | 4 | 1 | 1 | 3 | 2 | 1 | 3 | 1 | 1 | 2 |
dataset.dtypes()
Sample code number int64
Clump Thickness int64
Uniformity of Cell Size int64
Uniformity of Cell Shape int64
Marginal Adhesion int64
Single Epithelial Cell Size int64
Bare Nuclei object
Bland Chromatin int64
Normal Nucleoli int64
Mitoses int64
Class int64
dtype: object
target = ["Class"]
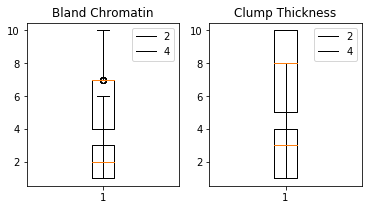
Method: boxplot
dataset.boxplot(features=["Bland Chromatin", "Clump Thickness"],
target=target)
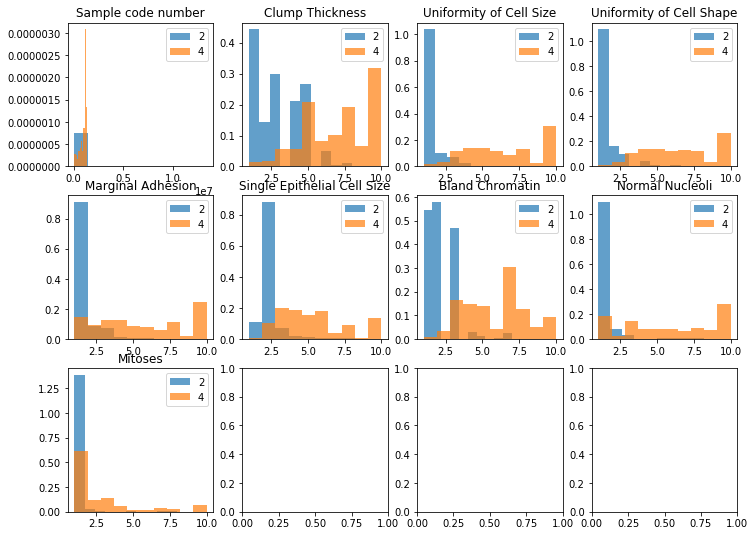
Method: dispersion_categoria
dataset.dispersion_categoria(target=target)
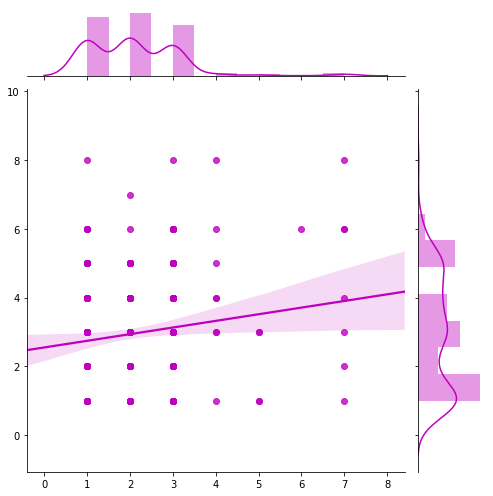
Method: sns_jointplot
dataset.sns_jointplot(feature1=["Bland Chromatin"], feature2=["Clump Thickness"], target=target,
categoria1 = [2])
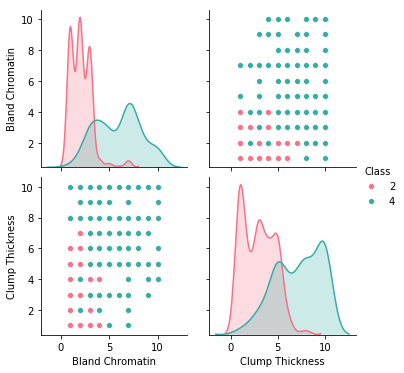
Method: sns_pairplot
dataset.sns_pairplot(features= ["Bland Chromatin", "Clump Thickness"], target=target)
Method: distribution_targets
dataset.distribution_targets(target=target)
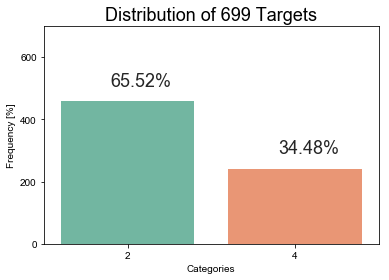
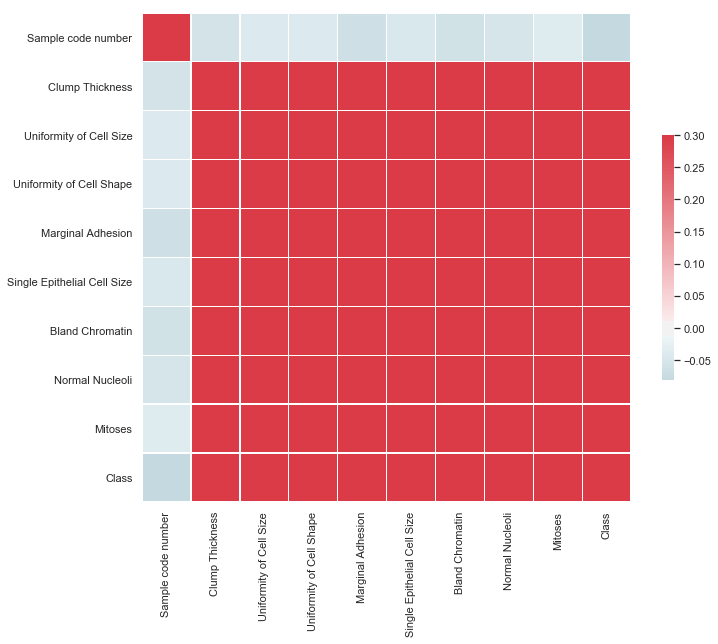
Method: corr_matrix
dataset.corr_matrix()
Method: not_type_object